Participação da professora de genética da Universidade Federal da Bahia (UFBA), Renata Ferreira de Lima.
segunda-feira, 9 de novembro de 2009
sábado, 17 de outubro de 2009
Reação em cadeia da polimerase - PCR

A técnica de PCR é um advento relativamente recente na história da biologia molecular, tendo sido desenvolvida nos anos 80 por Kary Mullis.
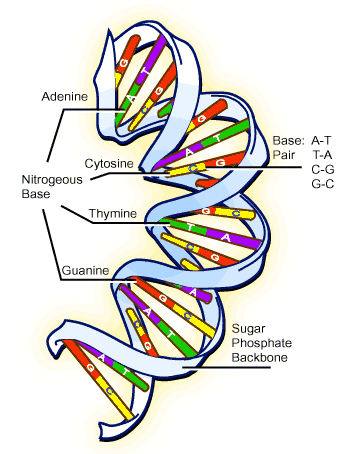
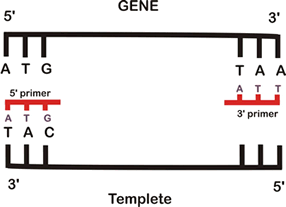
Na década de 1980, passou-se a utilizar a técnica do PCR para fazer milhares de cópias de um único pedaço de DNA. Essa técnica é usada em tubos de ensaio contendo o DNA e mais alguns compostos necessários, como primers (DNAs iniciadores) e a enzima DNA polimerase (enzima que faz a replicação do DNA). Os primers são fitas de DNA, com mais ou menos 20 bases (A, T, C, G) complementares, isto é se ligam por complementaridade ao início da seqüência de DNA que se quer multiplicar. Quando uma molécula de DNA vai ser multiplicada deve-se separar a dupla fita, formando assim duas fitas diferentes mas complementares entre si. Cada fita servirá de molde para a duplicação, por isso, precisamos de dois tipos de primers diferentes.
Veja as figuras clicando em figura 1 e figura 2.
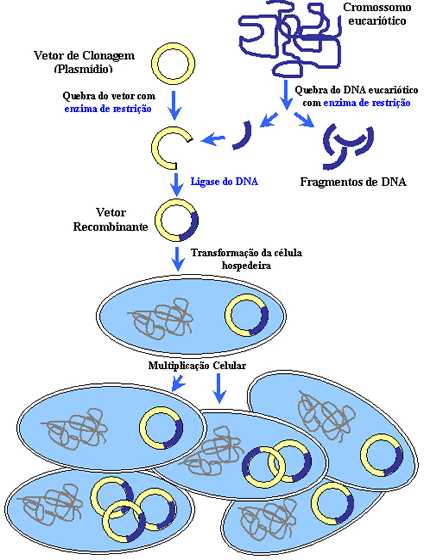
Passo a passo do processo da inserção do DNA num plasmídio
- Os pesquisadores querem estudar um gene humano que produz uma proteína que não se sabe a função.
- Os pesquisadores “recortam” (utilizando enzimas de restrição), do DNA humano, o gene de interesse.
- Esse fragmento de DNA contendo o gene é multiplicado por PCR para obtermos várias cópias do mesmo fragmento (ou da mesma informação).
- A mesma enzima que clivou o gene do DNA humano é utilizado para clivar o plasmídio bacteriano. Lembre-se que o fragmento de DNA, ao ser clivado, gera pontas adesivas que são complementares ao plasmídio se este for clivado com a mesma enzima.
- A seguir o plasmídio clivado é misturado com os fragmentos de DNA (contendo o gene) e uma enzima chamada ligase “cola” os fragmentos ao plasmídio, produzindo o chamado DNA recombinante. Isso feito, o DNA recombinante é introduzido em uma bactéria hospedeira.
- A bactéria hospedeira é colocada em um meio nutritivo seletivo, apenas aquelas que possuem o DNA recombinante crescem, formando colônias. Após muitas gerações de bactérias, o produto da expressão dos genes, as proteínas humanas, são purificadas das bactérias (são separadas das proteínas das bactérias).
Esse método produz uma grande quantidade de proteínas humanas possibilitando assim, seu estudo.
A tecnologia do DNA recombinante
Cada fragmento de DNA, que foi clivado e separado do resto do material genético, contém um ou mais genes. Lembre-se que cada gene origina uma proteína, portanto ao estudarmos o gene estamos estudando a proteína que ele codifica.
Mas o que devemos fazer para estudar o gene?
Devemos introduzi-lo no material genético (no DNA) de um hospedeiro para que ocorra a transcrição do gene, em mRNA, e a tradução em proteína.
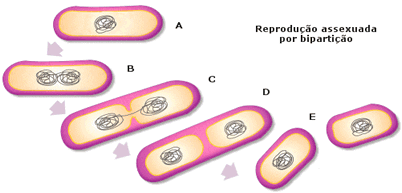
O hospedeiro é um organismo que se multiplica (se reproduz) rapidamente, como por exemplo, as bactérias. Quando as bactérias se reproduzem por bipartição elas transmitem ao seus “filhos” o seu material genético, portanto se neste material conter o fragmento de DNA de estudo, em pouco tempo teremos milhões de bactérias com o gene.
Clique aqui para ver a imagem.
O plasmídio é o matérial genético circular não ligado ao cromossomo que fica espalhado pelo hialoplasma das bactérias. Ele sofre o mesmo processo do DNA cromossomal de transcrição e tradução, além de, se multiplicar a cada divisão celular, passando uma cópia para cada célula “filha”.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
O plasmídio é retirado das células bacterianas para que se possa inserir o gene de estudo, para depois recoloca-lo na bactéria.
Inserção de DNA estranho num plasmídio. Clique aqui.
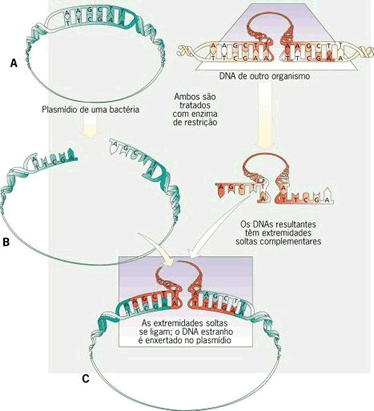{kind=link}
Técnica de PCR, passo a passo:
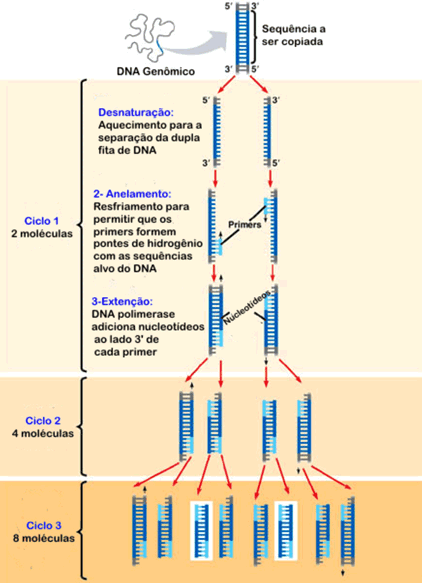
- A amostra de DNA, a enzima que faz a replicação (DNA polimerase), os nucleotídeos de DNA e os primers complementares a seqüência de DNA são colocados em um tubo de ensaio.
- Coloca-se o tubo de ensaio em uma máquina de PCR (maquina que aumenta e diminui a temperatura de acordo com um programa). Os passos seguintes, de aquecimento e resfrimento, acontecem dentro da máquina controlados pelo programa.
- Aquece-se o tubo a 94ºC para desnaturar (separar a dupla fita) o DNA.
- Cada fita simples do DNA que foi desnaturado serve de molde para a síntese de novas cadeias complementares. Para isso resfria-se a 54ºC onde os primers se anelam ao início das duas fitas simples, servindo de iniciadores para a enzima polimerase.
- Aquece-se novamente o tubo a 72ºC (temperatura ideal de funcionamento da DNA polimerase) para a duplicação da fita. A DNA polimerase inicia, após o final do primer, a colocar os nucleotídeos livres na fita de DNA ligando-os por complementariendade, formando assim uma nova fita dupla.
Assinar:
Comentários (Atom)